
Cardiomyopathie Hypertrophique (CMH)
Il s'agit d'un épaississement du myocarde ou muscle cardiaque ventriculaire, ce qui l'empêche, au fur et à mesure, de remplir son travail de pompe. Il ne s'agit pas d'une maladie cardiaque mais un syndrome, commun à de nombreuses maladies (cardiaques ou non). Certaines formes peuvent être d'origine génétique.
Ce syndrome évolue de façon variable selon les individus entre quelques mois et plus de quinze ans. Il touche toutes les races de chat, y compris les chats communs. Certaines races sont plus exposées : Maine Coon, Ragdoll et Sphynx.
I. Les causes de la CMH :
Toutes les causes ne sont pas identifiées. La plupart des CMH sont les conséquences d'une maladie générale comme : hypertension, insuffisance rénale chronique, hyperthyroïdie. En cas de suspicion de CMH, il faut donc commencer par rechercher une maladie générale.
I.1 Les formes génétiques :
Elles ne sont pas aussi fréquentes que l'on pourrait le penser et seules certaines mutations dans certaines races sont à ce jour connues. Chez le Maine Coon et le Ragdoll, on a pu identifier une mutation de CMH 1 au sein du gène MyBPC3 *. Cette mutation modifie la séquence du gène qui code une protéine (la protéine C) qui participe à la contraction du muscle cardiaque. Le corps ne produit alors plus de codon GCC ** mais le CCC ***. Les protéines ne peuvent plus agir correctement lors de la contraction du muscle cardiaque. Pour compenser, le corps produit plus de protéines pour contracter le muscle, mais seule environ la moitié de ces codons ne sont pas mutés. Ceci entraîne un épaississement des parois du coeur. L'allèle muté est alors dominant.
I.2 Mode de transmission :
Pour une forme génétique une transmission héréditaire existe. Les modalités seront variables selon le type de mutation. On peut trouver des familles entières atteinte du CMH. La maladie touche donc autant les mâles que les femelles et un seul des parents suffit à transmettre la maladie à sa descendance. Chez le Ragdoll, le Persan ou l'American Shorthair, elle est beaucoup moins présente car il s'agit d'un gène récessif.
II. Les symptômes :
Certains animaux pouvant, bien qu'atteint d'une forme grave, ne présentent aucun symptôme et aucune anomalie lors de l'auscultation cardiaque. Cette situation explique un certain nombre de morts subites rencontrées dans l'espèce féline.
- fatigue rapide lors d'exercice physique
- insuffisance cardiaque (arythmie, tachycardie, souffle au coeur)
- embolie ou œdème pulmonaire
- difficultés à respirer
- paralysie des nombres arrières secondaires à l'embolie d'un caillot (ou thrombose) qui se forme généralement dans l'oreillette gauche très anormalement dilatée
Le rythme cardiaque peut être tout à fait normal. Il arrive toutefois régulièrement qu'aucun des symptômes n'ait été remarqué lorsque survient la mort du chat.
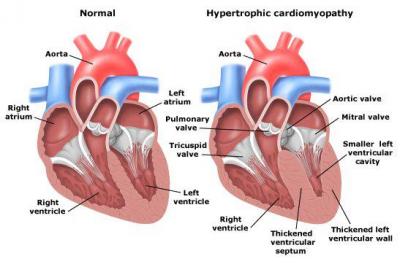
III. Dépistage :
L'examen de choix pour identifier une hypertrophie du myocarde est actuellement l'échographie cardiaque. Si il y a confirmation de la présence d'un CMH, il faut rechercher une cause éventuelle, par des examens sanguins et d'une mesure de la pression artérielle.
Les races Maine Coon et Ragdoll sont les races les plus suivies pour la maladie. La recherche a permis d'identifier une mutation génétique propre au Maine Coon et au Ragdoll, décelable par test ADN. Ce test permet de détecter la mutation du gène MyBPC3 chez ces deux races. Il existe au moins une autre cause de CMH chez le chat, le test ADN n'est pas suffisant. Il faut compléter par une échographie des l'âge de 18 mois. Une échographie 2d et M-mode permettent de mesurer l'épaisseur des parois du coeur et la taille des cavités auxquelles on ajoute une échographie doppler couleur.
Il n'y a pas de traitement spécifique de l'hypertrophie du myocarde. Un traitement approprié est décidé quand une cause est identifiée : traitement insuffisance rénale, hyperthyroïdie.
Dans le cas d'une insuffisance cardiaque, un traitement visant à améliorer la qualité de vie de l'animal et à prévenir d'éventuelles complications.
Polymorphisme de ce syndrome : âge variable de manifestation, degré variable d'évolution et d'expression, symptômes variables. Ce gène voit son activité modulée par d'autres gènes et/ou d'autres facteurs externes, épigénétiques. Il existe un certain nombre d'interrupteurs ou de potentiomètres qui accélèrent ou retardent le réveil du gène ainsi que l'intensité de son expression.
L'existence d'une autre cause génétique au moins, chez le Maine Coon est admise : CMH 2. Des recherches sont en cours. Combien d'autres mutations causatives? De formes congénitales et non héréditaires?
L'utilisation du test ADN et un suivi par échographie des reproducteurs, doit permettre de réduire la transmission de cette forme connue. L'échographie cardiaque n'a aucune valeur de pronostic quant à la santé cardiaque future d'un chat. Elle ne peut indiquer que l'état du coeur au moment où est pratiqué l'examen. Elle garde toute son importance dans le suivi des chats qui ne sont pas porteurs de la mutation MyBPC3, afin de dépister des CMH ayant d'autres causes.
Le test ADN permet d'avoir une certitude : tout chat qui a la mutation, qu'elle soit à l'etat homozygote ou hétérozygote, est un chat qui développera tôt ou tard la pathologie du moins dans la majorité des cas. Ils sont nombreux les coons dépistés génétiquement "CMH 1 hétérozygote" qui avaient ou ont encore un beau coeur à l'échographie.
Il est indispensable de ne pas faire de raccourcis : l'élevage est d'accepter d'être le garant de l'amélioration d'une race et non pas de son appauvrissement.
Sources : pawpeds/antagéne/Kittleson et Meurs/cooncept/recherches personnelles
Rédigé par la Baie des Coons
(*) myosin-binding protein C
(**) guanine-cytosine-cytosine
(***) protéine C-cytosine-cytosine
Codon : séquence de trois nucléotides sur un acide ribonucléique messager (ARNm) spécifiant l'un des 22 acides aminés protéinogènes dont la succession sur l'ARN messager détermine la structure primaire de la protéine à synthétiser.